Whole-exome sequencing identifies a GREB1L variant in a three-generation family with Müllerian and renal agenesis: a novel candidate gene in Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome. A case report
Project Description
-
Explored the genetic cause of Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, a rare disorder characterized by uterovaginal agenesis in 46,XX women.
-
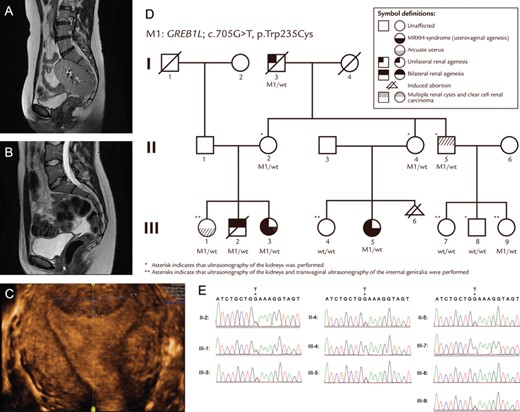
Investigated a unique three-generation family with two female members affected by MRKH and unilateral renal agenesis (RA), along with two deceased male relatives with RA.
-
Applied whole-exome sequencing (WES) on eight family members to identify a novel pathogenic variant (c.705G>T) in the GREB1L gene.
-
Provided strong evidence supporting GREB1L as a key gene in kidney and female reproductive tract development.
-
The family’s genetic pattern suggests autosomal dominant inheritance with incomplete penetrance, possibly influenced by genomic imprinting.
-
This is the first WES-based study on a large MRKH family pedigree, highlighting GREB1L as a promising candidate gene in understanding MRKH syndrome.
Project Details
Results
-
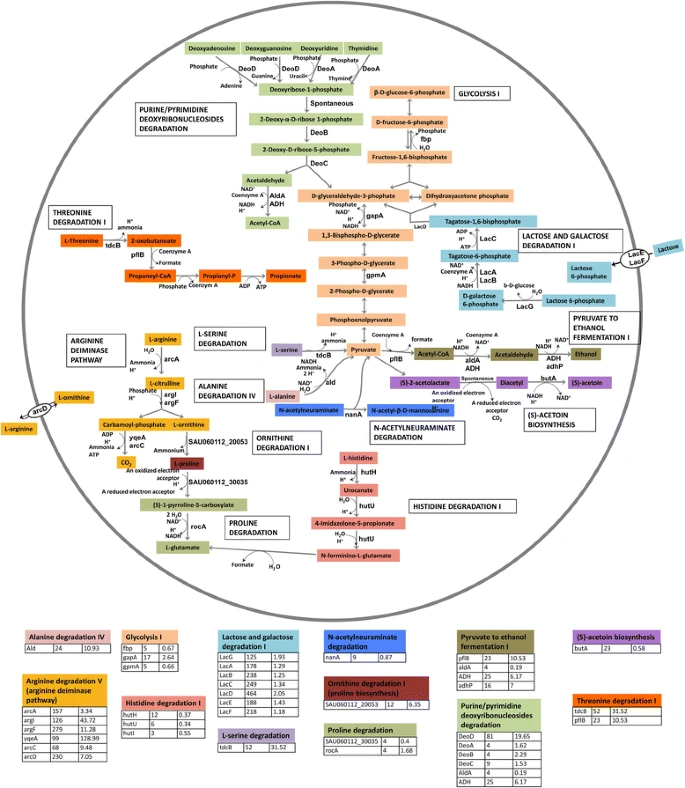
Identified over 131,000 unique variants through whole-exome sequencing (WES) across eight family members.
-
Narrowed down to three shared variants, with one novel heterozygous missense variant in the GREB1L gene (c.705G>T) identified as the strongest candidate.
-
This variant:
-
Alters a highly conserved amino acid (tryptophan to cysteine).
-
Predicted to disrupt protein structure and splicing signals.
-
Scored 33 on the CADD scale (top ~0.05% of pathogenic potential genome-wide).
-
Rated as deleterious by SIFT, PolyPhen, and PROVEAN tools.
-
-
GREB1L is known to play a critical role in kidney and reproductive tract development, aligning with the family’s clinical features.
-
Sanger sequencing confirmed the variant in additional family members, supporting its segregation with the phenotype.
-
Inheritance pattern shows incomplete penetrance and possible parent-origin-specific expression (imprinting).
-
Two other variants (in DSG3 and PGK2) were deemed not biologically relevant to MRKH syndrome.
-
No large deletions or duplications detected in key chromosomal regions (17q12, 22q11, 16p11.2).
-
Additional analysis found no genetic cause for renal cysts in one affected uncle, suggesting a distinct etiology for his case.
Goals Achivied
-
Identified a novel disease-associated variant: Discovered a previously unreported missense variant (c.705G>T) in the GREB1L gene, which is strongly associated with MRKH syndrome type II and renal agenesis.
-
Strengthened genetic understanding of MRKH syndrome: Provided compelling evidence that GREB1L is a strong candidate gene contributing to the genetic basis of MRKH type II, especially in familial cases.
-
Revealed a potential parent-origin effect: Uncovered a parent-origin-specific pattern of inheritance, suggesting genomic imprinting may influence GREB1L variant penetrance—an insight not previously well-documented in MRKH research.
-
Validated through extended family analysis: Used whole-exome sequencing (WES) and segregation analysis across three generations, supporting a pattern of autosomal dominant inheritance with incomplete penetrance.
-
Contributed to future diagnostic pathways: Positioned GREB1L as a potential biomarker for genetic testing in suspected familial MRKH syndrome cases, especially those with co-occurring renal anomalies.
Explore the full research here